Analytics engineering is creating and disseminating knowledge within organizations
dbt Labsの創業者であり、analytics engineeringという概念のパイオニアであるTristan Handyのページがある。
彼はここでanalytics engineeringを以下のように定義している。
creating and disseminating knowledge within organizations
私にとってのanalytics engineeringといえば、データのユースケースを見つけ、データモデリングをして、エンドユーザーに必要なデータを届ける仕事という、実装寄りの理解だった。 しかし、上記の表現は、より目的が明確で今の感覚にフィットする。
これに加えて自分なりに要素を足すなら、人と人、人とデータを繋ぐCatalystとしての側面を強調したい。もちろんTristanの定義する disseminating にはそのニュアンスも含まれていると思うが、単に仕組みを作るだけでなく、間に入って反応を促進させる触媒という意識は、現場で動く上で常に持っていたい。
How to Lead in Data Science
最近、以前読んだデータサイエンスのマネジメント本を改めて読み直した。 データチームを率いる立場として、今の自分自身の課題感やチームの方針とリンクする部分が多い。簡単に備忘録として残しておく。
日本語版も最近出た:
- Chapter 1に出てくるcaseがかなりリアル。後のchapterで、それぞれのcaseに対する処方箋が載っており、自分だけでなくチームメンバーを導くケースでも役に立ちそう。
- Data scienceで活躍するにはデータ分析の手法とかに明るいだけではいけないのはよく思っているが、そのあるべきを明快に言語化してくれているように思った。特にChapter 6のtechnology readmapを描きそれをexecuteしていくべきという話や、sponsor, championを明確にしプロジェクトを進めるべきという話は、自分がレベルアップする上で必要だなと感じている。
- ではどういうロードマップを描くべきかという点で、Chapter 6には8つも具体的なroadmapのタイプを書いており、参考になる。
- 総じて抽象と具体の行き来が上手な文章であり、良い文章を書くにはという点でも勉強になる。
RでERGMを実装してみた
ネットワーク分析に明るい方であれば、2019年に書かれたこの野心的な記事を覚えている人も多いのではないだろうか。
statnet チームが提供している R のパッケージに頼らずに、Exponential Random Graph Model (ERGM) をできるだけスクラッチで実装しようという壮大な試みである。
実装した関数による推定値は、残念ながら {ergm} で推定した値と乖離してしまったという結びになっている。
その原因解明は多くの読者にとって待ち望んでいたことに違いない。
そこで時間を持て余した筆者が、ゴールデンウィークを使って ERGM を (MPLE を除いて) 一から、かつ {Rcpp} を用いて高速に推定できるよう実装した。*1
以下の repository にコードをパッケージとして公開しているので、興味のある方は遊んでみていただけると幸いである。
flomariagge ~ edges + triangle という単純なモデルを {myergm}、{ergm} で 100 回ずつ推定し、その推定値の分布をプロットしたものが以下の図である。
両者は概ね一致しているように思われる。


先の記事ではなぜ推定がうまくいかなかったのか?
気になるのが、一番最初にあげた記事でなぜ ERGM の推定値が {ergm} のそれと比較して大きく異なってしまったのかである。
その原因が、以下の関数にあることを特定した。
#対数尤度比を計算する関数 loglikelihood_ratio <- function(theta,fit,n){ sample_length <- length(fit$edges) sample_index <- (sample_length-n):sample_length sampled_edge_count <- fit$edges[sample_index] sampled_triangle_count <- fit$triangle[sample_index] theta0 <- c(fit$theta0_edges,fit$theta0_triangle) g_obs <- c(count_edges(fit$obs_graph),count_triangle(fit$obs_graph)) first = (theta - theta0) %*% g_obs exp_vec <- c() for(i in 1:n){ sam <- exp((theta - theta0) %*% c(sampled_edge_count[i],sampled_triangle_count[i])) exp_vec <- c(exp_vec,sam) } second <- log(sum(exp_vec)/length(exp_vec)) loglikelihood_ratio <- first - second return(loglikelihood_ratio) }
sample_index <- (sample_length-n):sample_length とあるので、log likelihood ratio を計算するときに、MCMC で生成されたネットワークの数を としたとき、
番目から
番目までの連続した全てのサンプルを使っていることになる。
一方、{ergm} には control.ergm() という関数があり、MCMC をチューニングできるが、そのデフォルトは以下のようになっている。
MCMC.interval = 1024, MCMC.burnin = MCMC.interval * 16, MCMC.samplesize = 1024,
まず、1024 * 16 を burnin とする。
その後、MCMC で生成されたグラフ同士の間隔が 1024 となるように (MCMC.interval = 1024)、1024 個のグラフをサンプリングする (MCMC.samplesize = 1024)。
そのサンプリングされた 1024 個のグラフを推定値を得るために用いるのである。
これを踏まえると、最初の記事で推定がうまくいかなかった原因は、MCMC でサンプリングされたグラフ同士の間隔が 0 であり、グラフ同士の serial correlation が強いことに原因がありそうである。
そこで、{myergm} でグラフ同士の相関が強く残るようにサンプリングして推定した場合の推定値の分布を見てみよう。
推定がうまくいかなかったケースを、仮に arabou_ergm と呼ぶことにすると、それは以下のように実装できる。
# Number of simulations R <- 100 df <- foreach(i = 1:R, .combine = "rbind") %do% { # Estimate the parameters. theta_arabou_ergm <- myergm::myergm_MCMLE(model = flomarriage ~ edges + triangle, seed = i, p_one_node_swap = 0, p_large_step = 0, p_invert = 0, MCMC_samplesize = 1024, MCMC_interval = 1, second_round = FALSE) # Return the output. return(data.frame("iter" = i,"param" = c("edges", "triangle"), "arabou_ergm" = theta_arabou_ergm)) }
{ergm} で推定したケースと比較したとき、それぞれの分布は以下の通りになる。


arabou_ergm の方が推定値にかなりばらつきが出ていることがわかる。
私が実装した {myergm} は、厳密には {ergm} の実装とは異なる点がある。
例えば、{ergm} の MCMC のデフォルトの sampler は "tie-no-tie" といって、最初にリンクが存在する dyad か、リンクが存在しない dyad どちらの状態を変化させるかをそれぞれ確率 1/2 で決定し、リンクを加える/消す。
一方 {myergm} では、tie-no-tie sampler は実装しておらず、代わりに Mele (2017) が提案したような large step を許容した sampler、つまりある確率で 1 回のステップで複数の dyad の状態を変化させるような実装になっている。
ただし、これらの MCMC sampling の手法は推定値に大きく影響を与えるものではなく、大事なのは推定に用いるグラフの sampling の方であったようだ。 他にも ERGM には複雑怪奇なことが多いので*2、それらは今後の検討課題としたい。
References
- Hummel, R. M., Hunter, D. R., & Handcock, M. S. (2012). Improving simulation-based algorithms for fitting ERGMs. Journal of Computational and Graphical Statistics, 21(4), 920-939.
- Hunter, D. R., Goodreau, S. M., & Handcock, M. S. (2013). ergm. userterms: A Template Package for Extending statnet. Journal of statistical software, 52(2), i02.
- Mele, A. (2017). A structural model of dense network formation. Econometrica, 85(3), 825-850.
Rで疎行列に親しむ
仕事でネットワーク分析に苦しむ機会が増えてきた。
その中で疎行列 (sparse matrix) を取り扱う重要性が高まってきている。
本稿の目的は疎行列の意義と使い方を学び、仲良くなることである。

疎行列とは
疎行列とは、要素のほとんどが 0 であるような行列のことである。
巨大な行列を用いた計算が必要になることがある。 行列が巨大になるときに問題となるのは、行列がメモリを圧迫することである。
例えば、1万×1万の単位行列を、以下のように適当に作成する。
I <- diag(nrow = 10000, ncol = 10000) pryr::object_size(I)
800 MB
単純な単位行列ではあるが、800 MB ものメモリが割り当てられている。
これが、100万×100万の単位行列を普通に作成すると、以下の通りになる。
I <- diag(nrow = 1000000, ncol = 1000000)
Error: cannot allocate vector of size 7450.6 Gb
たった1つの単位行列を作成するために、7 TB ものメモリーが必要となってしまう。 この行列のほとんどは 0 であるが、それらがわざわざ全て保存される結果、このように巨大な容量を食ってしまう。
ということは、行列の要素がほとんど 0 であるという特性を活かせば、無理なく作成することができるはずである。
そこで疎行列の出番である。RではMatrixパッケージを用いて以下のように作成できる。
I_sp <- Matrix::sparseMatrix(i = 1:1000000, j = 1:1000000) I_sp[1:3, 1:3]
3 x 3 sparse Matrix of class "ngCMatrix"
[1,] | . .
[2,] . | .
[3,] . . |
pryr::object_size(I_sp)
8 MB
100万×100万の単位行列も、わずか8MBで作成できる。 対角要素は全てbooleanで表現されているので、そのおかげでさらにメモリ容量が節約できている。
I_sp[1, 1]
[1] TRUE
これを全て1にすると以下の通りとなり、メモリへの割当が多少増加する。
I_one <- as(I_sp, "dgCMatrix") I_one[1, 1]
[1] 1
pryr::object_size(I_one)
16 MB
行列演算も通常と同じようにできる。 面白くない例だが、疎行列の単位行列を2つ掛け合わせることは以下のようにできる。
I_sp %*% I_sp
ネットワーク分析における疎行列
グラフで誰と誰がつながっているかを表現するために使われる行列を、隣接行列 (adjacency matrix) と呼ぶ。
例えばノードの数が3つで、1 -- 2 -- 3 のようなつながりのある無向グラフは
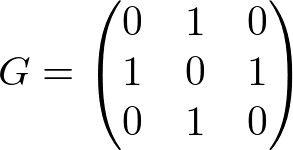
と表現できる。つまりGの(i, j)要素は、ノードiとjの繋がりを表現する。
グラフが小さいときはまだ上のように表現できる。 しかしグラフが巨大になるとメモリを圧迫してしまうのは、上で見た例の通りである。 そこで、多くのグラフは sparse であるという特性を活かし、グラフを疎行列で表現することがよく使われる。
Rでネットワーク分析を行うときによく使われるパッケージ群の1つとしてstatnetがある。
例えば、指数ランダムグラフモデル (exponential random graph model; ERGM) を推定するときは、
networkでネットワークオブジェクトを作成して、それに基づき推定を行う。
以下はその一例である。
library(statnet) data(florentine) est <- ergm(formula = flomarriage ~ edges + absdiff("wealth") + triangle)
しかしそのネットワークに色々と操作を加えるときはそのネットワークを一度隣接行列に置き換えて処理を行う必要が出てくる。 その際にネットワークオブジェクトを隣接行列に置き換えるのは、例えば以下のようなコードがある。
adj <- network::as.matrix.network.adjacency(flomarriage) head(adj[, 1:3])
Acciaiuoli Albizzi Barbadori Acciaiuoli 0 0 0 Albizzi 0 0 0 Barbadori 0 0 0 Bischeri 0 0 0 Castellani 0 0 1 Ginori 0 1 0
しかしどういうわけか、network::as.matrix.network.adjacencyは密行列 (dense matrix) して隣接行列を返す。
これはネットワークが大きい場合、メモリを大きく圧迫してしまう。
それを回避するためには、まずネットワークをedgelist、つまりつながっているノード同士だけを取り出す。
edgelist <- network::as.edgelist(flomarriage)
そこからこのネットワークを疎行列として表現する。
adj_sp <- Matrix::sparseMatrix(i = edgelist[, 1], j = edgelist[, 2], symmetric = TRUE)
pryr::object_size(adj)
3.58 kB
pryr::object_size(adj_sp)
1.82 kB
小さいネットワークだと対した差ではないが、これがノード数が増えると両者の差は爆発的に広がっていく。
Rcppで疎行列をたしなむ
隣接行列を用いた重たい処理が必要なときは、Rの疎行列をRcppに渡して高速化することができる。
ここではRcppArmadilloを用いてやってみる。 やったことはないが、RcppEigenでも同じようにできるらしい。
その処理の例として、ノードiとjのつながりのうち、iとjが同じ特徴量を持つような隣接行列を作ることを考える。 例えば特徴量として性別を考えたとき、同じ性別同士で実際につながっているリンクのみ含んだ隣接行列を作成したい。
まずは適当にグラフを作成し、疎行列で表現する。
# This seed comes from the result of the 2020 Hawks-Giants Japan Series. set.seed(264) # Number of nodes N <- 1000 # Create an edgelist. I consider an undirected network. edgelist <- tibble::tibble(tail = 1:N, head = 1:N) %>% tidyr::expand(tail, head) %>% dplyr::filter(tail < head) %>% dplyr::mutate(connected = as.integer(unlist(purrr::rbernoulli(n = nrow(.), p = 0.01)))) %>% dplyr::filter(connected == 1) # Create a sparse adjacency matrix net <- Matrix::sparseMatrix(i = edgelist$tail, j = edgelist$head, dims = c(N, N), symmetric = TRUE) net <- as(object = net, Class = "dsCMatrix")
次にノードの特徴量を適当に作成する。
# Vertex feature like gender. x <- unlist(purrr::rbernoulli(n = N))
そして今やりたい処理をC++のコードで書く。
RcppArmadilloで疎行列を扱うときはarma::sp_matを用いる。
#include <RcppArmadillo.h> // [[Rcpp::depends(RcppArmadillo)]] // [[Rcpp::export]] arma::sp_mat get_connected_same_feature_dyads_cpp(const arma::sp_mat& adjmat, const arma::vec& feature) { int n_nodes = adjmat.n_rows; arma::sp_mat output(n_nodes, n_nodes); for (int i = 0; i < n_nodes; i++) { for (int j = 0; j < n_nodes; j++) { if (i != j) { if (adjmat(i, j) == 1 & feature[i] == feature[j]) { output(i, j) = 1; } } } } return output; }
実際に実行してみる。
Rcpp::sourceCpp("src/get_connected_same_feature_dyads_cpp.cpp") output <- get_connected_same_feature_dyads_cpp(net, x)
net[1, 996] == 1 & x[1] == x[996]
[1] TRUE
output[1, 996]
[1] 1
正しく計算できているように思う。
別にやる必要はないが、同じ処理をRで実装すると以下の通りとなる。
get_connected_same_feature_dyads_R <- function(adjmat, x) { n_nodes <- length(x) output <- Matrix::sparseMatrix(i = {}, j = {}, dims = c(length(x), length(x))) for (i in 1:n_nodes) { for (j in 1:n_nodes) { if (i != j) { if (adjmat[i, j] == 1 & x[i] == x[j]) { output[i, j] <- TRUE } } } } return(output) }
ベンチマークを行うとRcppの方が圧倒的に速い。Rでforを書いた時点で負け確定であるのは内緒である。
time <- rbenchmark::benchmark( "R" = { output <- get_connected_same_feature_dyads_R(net, x) }, "cpp" = { output <- get_connected_same_feature_dyads_cpp(net, x) }, replications = 1 ) time
test replications elapsed relative user.self sys.self user.child sys.child 2 cpp 1 0.016 1.00 0.016 0.000 0 0 1 R 1 136.508 8531.75 136.516 0.004 0 0
Rを使ってより高速に処理するには一工夫必要なことも多いが、変に凝ったコードを書くぐらいならRcppで殴ってしまった方が良い。
終わりに
ぜひみなさんも疎行列に親しみ、ネットワーク分析に苦しみましょう。
broomを使って回帰分析の結果を出力する
いわずもがなであるが、『効果検証入門』は大変勉強になる。 因果推論は大学・大学院のときに学んでいるが、その復習に使える。 また、Rを使ってどのように因果推論を行うかについても有益な情報を与える。

その中で、回帰分析の結果の出力方法について私が知らないやり方があったので、ここで書いてみる。
それがbroomというパッケージを使用した方法である。
通常Rで線形回帰分析を行うときはlm関数を用いる。
例のごとく、irisを用いて適当な線形回帰モデルを推定し、その結果をsummaryで出力する。
reg <- lm(Sepal.Length ~ Sepal.Width + Petal.Length + Petal.Width, data = iris) summary(reg)
Call:
lm(formula = Sepal.Length ~ Sepal.Width + Petal.Length + Petal.Width,
data = iris)
Residuals:
Min 1Q Median 3Q Max
-0.82816 -0.21989 0.01875 0.19709 0.84570
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 1.85600 0.25078 7.401 9.85e-12 ***
Sepal.Width 0.65084 0.06665 9.765 < 2e-16 ***
Petal.Length 0.70913 0.05672 12.502 < 2e-16 ***
Petal.Width -0.55648 0.12755 -4.363 2.41e-05 ***
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
Residual standard error: 0.3145 on 146 degrees of freedom
Multiple R-squared: 0.8586, Adjusted R-squared: 0.8557
F-statistic: 295.5 on 3 and 146 DF, p-value: < 2.2e-16
この方法では、下に余計な情報が出てくるなど見づらい、kableExtraなどで出力できないなど不便なことが多い。
そこでbroom::tidyの出番である。
推定された係数の情報のみを抽出し、tibbleとして出力してくれる。
reg <- lm(Sepal.Length ~ Sepal.Width + Petal.Length + Petal.Width, data = iris) %>% broom::tidy()
# A tibble: 4 x 5 term estimate std.error statistic p.value <chr> <dbl> <dbl> <dbl> <dbl> 1 (Intercept) 1.86 0.251 7.40 9.85e-12 2 Sepal.Width 0.651 0.0666 9.77 1.20e-17 3 Petal.Length 0.709 0.0567 12.5 7.66e-25 4 Petal.Width -0.556 0.128 -4.36 2.41e- 5
大概の場合、関心のあるのは係数の推定値のみであるから、これでほとんど事足りる。
加えてtibbleとして出力してくれるので、例えばkableExtra::kable(reg)として出力することができる。
論文への出力のためのtableはstargazerを用いることが多いが、書き方によってはこのbroomを用いることで置き換えることが可能かもしれない。
purrrを使ってみる
Rのパッケージpurrrは、リスト/ベクトルに対して反復処理を行う関数を提供するパッケージである。
これどう呼ぶのが正しいのだろうか。ひとまず「ぴゅるるる」と呼ぼう。
Rでループ処理を行うときは、処理速度が極端に遅いため極力forを使わないようにすべしというのは、ある程度Rを使っている方であればご存知かと思う。
他にはapplyもあるが、個人的にあまり使いやすさを感じたことはない。
purrrは、速度そして可読性の両方を兼ね備えたパッケージである。
基本的な使い方
purrrのメインとなる関数はpurrr::mapである。purrr::map(x, f)として、リスト/ベクトル/行列/データフレームxに対して関数fを繰り返し作用させ、計算結果をリストに格納する。
基本的にはx %>% purrr::map(f)のように、リスト/ベクトルを渡して関数で処理するというパイプラインにするのが一般的かと思う。
例えば、標準正規分布から10個の乱数を取ってきて、それらを5つのリストに格納するコードは以下のようにかける。
library(magrittr) 1:5 %>% purrr::map(rnorm, n = 5)
[[1]] [1] 1.2644874 -0.1058268 1.1207076 0.9976939 0.1906772 [[2]] [1] 0.3033158 1.4178929 2.4092054 1.6336716 2.7202050 [[3]] [1] 2.293005 3.507343 3.918410 2.050489 2.193373 [[4]] [1] 3.697953 4.015712 5.237348 4.090736 4.329179 [[5]] [1] 6.763691 4.920744 4.854612 4.336114 5.076095
私の場合、このpurrrを使うシチュエーションは主に2つある。
1. 同じグラフをそれぞれのカテゴリー/グループに対してサクッと作成し、サクッと取り出せるようにしたいとき
1つ目は、同じ種類のグラフ (ヒストグラム、散布図など) を、違うカテゴリー/グループごとに作成したいときである。
ggplot2のfacet_gridなどを使って、1つの図にまとめる方法もあるが、カテゴリーの数が多すぎるとグラフが小さくなり問題である。
自分の好きなカテゴリーのグラフを手軽に取り出したいときもある。
そこでpurrrの出番である。
irisデータを用いて、アヤメの種類ごとガクの長さ (sepal length) のヒストグラムを作成することを考える。
library(ggplot2) draw_hist <- function(df, column) { g <- ggplot(data = df, aes(x = !!dplyr::enquo(column))) + geom_histogram() return(g) } hist <- iris %>% split(.$Species) %>% purrr::map(draw_hist, column = Sepal.Length)
こうすることで、それぞれのアヤメの種類ごとにガクの長さを、例えばhist$setosaのようにして、すぐにsetosaのヒストグラムを呼び出すことができる。

もっというと、irisをlong形式に変換し、各カテゴリー・各変数ごとにsplitしてpurrr::mapしてしまえば、全てのグループと変数の組み合わせについてヒストグラムを作成することができる。
hist_all <- tibble::rowid_to_column(iris, var = "id") %>% tidyr::pivot_longer(-c(id, Species), names_to = "variable", values_to = "value") %>% split(list(.$Species, .$variable)) %>% purrr::map(draw_hist, column = value) hist_all$setosa.Petal.Width

データサイズが大きくなったり、分割するカテゴリーが増えたりするとメモリー不足にならないか心配であるが、とりあえずこれまではうまく動いている。
ちなみに上の自作関数の中にある!!dplyr::enquoはnon-standard evaluation (NSE) と呼ばれる黒魔術で、今は無視してほしい。*1
2. 違うデータフレームを続けて結合したいとき
2つ目は、あるデータセットに対して、複数の異なるデータセットを次々と結合させたいときである。 例えば、上で使用したアヤメのデータセットが、アヤメの種類 (Species) 、ガクの長さ (Sepal.Length) 、ガクの幅 (Sepal.Width) 、花弁の長さ (Petal.Length )、花弁の幅 (Petal.Width) の5つに分割されており、これらは全てあるid変数で結合できるとしよう。
df_iris <- tibble::rowid_to_column(iris, var = "id") df_species <- dplyr::select(df_iris, c(id, Species)) df_sepallength <- dplyr::select(df_iris, c(id, Sepal.Length)) df_sepalwidth <- dplyr::select(df_iris, c(id, Sepal.Width)) df_petallength <- dplyr::select(df_iris, c(id, Petal.Length)) df_petalwidth <- dplyr::select(df_iris, c(id, Petal.Width))
(このコードもpurrrでスマートに書けそうな気がするが、ひとまず力技で押し通した)
これらを元通りに結合するとき、1つのやり方は素直にdplyr::left_joinを繰り返し行うことだろう。
df <- dplyr::left_join(df_species, df_sepallength, by = "id") %>% dplyr::left_join(., df_sepalwidth, by = "id") %>% dplyr::left_join(., df_petallength, by = "id") %>% dplyr::left_join(., df_petalwidth, by = "id") head(df)
id Species Sepal.Length Sepal.Width Petal.Length Petal.Width 1 1 setosa 5.1 3.5 1.4 0.2 2 2 setosa 4.9 3.0 1.4 0.2 3 3 setosa 4.7 3.2 1.3 0.2 4 4 setosa 4.6 3.1 1.5 0.2 5 5 setosa 5.0 3.6 1.4 0.2 6 6 setosa 5.4 3.9 1.7 0.4
ただ、コードとしては冗長な感は否めない。どのデータフレームをjoinしたか、混乱することもしばしばある。
私も最近知ったのだが、これはpurrr::reduceを用いるとスッキリと書くことができる。
df <- list(df_species, df_sepallength, df_sepalwidth, df_petallength, df_petalwidth) %>% purrr::reduce(dplyr::left_join, by = "id") head(df)
id Species Sepal.Length Sepal.Width Petal.Length Petal.Width 1 1 setosa 5.1 3.5 1.4 0.2 2 2 setosa 4.9 3.0 1.4 0.2 3 3 setosa 4.7 3.2 1.3 0.2 4 4 setosa 4.6 3.1 1.5 0.2 5 5 setosa 5.0 3.6 1.4 0.2 6 6 setosa 5.4 3.9 1.7 0.4
繰り返しにはforeachもよい?
forは使うなと冒頭で述べたが、私はちょくちょくforeachを使う。並列処理もforeachで行うことができ、便利である。
標準正規分布から10個の乱数を取ってきて、それらを5つのリストに格納するコードは、foreachを用いて以下のようにも書ける。
library(foreach) foreach(i = 1:5) %do% { rnorm(n = 10) }
が、purrrを用いた方が処理速度は速い。それはそうか。
rbenchmark::benchmark( use_purrr = { 1:100 %>% purrr::map(rnorm, n = 1000) }, use_foreach = { foreach(i = 1:100) %do% { rnorm(n = 1000) } } )
test replications elapsed relative user.self sys.self 2 use_foreach 100 2.959 3.595 2.935 0.014 1 use_purrr 100 0.823 1.000 0.810 0.011
foreachについてはまた別のポストで書いてみたい。
特に並列処理を行うとき、なぜnamespace::functionと普段から書いておいたほうが良いかがわかる。
*1:いつも雰囲気で何とかしている。
dplyr::acrossを使ってみる
Rでdata frameの特定の列に対してある処理を施すとき、私はdplyr::mutate_atやdplyr::mutate_ifをよく使用している。
例えば、本来はnumeric型であるべき列がcharacter型となっているとき、それらの列を全てnumeric型に戻したいときがある。
以下は逆に、dplyr::mutate_ifを用いてnumeric型のものを全てcharacter型にする。
library(magrittr) iris %>% dplyr::mutate_if(.predicate = is.numeric, as.character) %>% tibble::as_tibble() # A tibble: 150 x 5 Sepal.Length Sepal.Width Petal.Length Petal.Width Species <chr> <chr> <chr> <chr> <fct> 1 5.1 3.5 1.4 0.2 setosa 2 4.9 3 1.4 0.2 setosa 3 4.7 3.2 1.3 0.2 setosa 4 4.6 3.1 1.5 0.2 setosa 5 5 3.6 1.4 0.2 setosa 6 5.4 3.9 1.7 0.4 setosa 7 4.6 3.4 1.4 0.3 setosa 8 5 3.4 1.5 0.2 setosa 9 4.4 2.9 1.4 0.2 setosa 10 4.9 3.1 1.5 0.1 setosa # … with 140 more rows
これを、dplyr::across関数を使うと、このように書ける。
iris %>% dplyr::mutate(dplyr::across(where(is.numeric), as.character)) %>% tibble::as_tibble() # A tibble: 150 x 5 Sepal.Length Sepal.Width Petal.Length Petal.Width Species <chr> <chr> <chr> <chr> <fct> 1 5.1 3.5 1.4 0.2 setosa 2 4.9 3 1.4 0.2 setosa 3 4.7 3.2 1.3 0.2 setosa 4 4.6 3.1 1.5 0.2 setosa 5 5 3.6 1.4 0.2 setosa 6 5.4 3.9 1.7 0.4 setosa 7 4.6 3.4 1.4 0.3 setosa 8 5 3.4 1.5 0.2 setosa 9 4.4 2.9 1.4 0.2 setosa 10 4.9 3.1 1.5 0.1 setosa # … with 140 more rows
dplyr::across関数の第1引数として列を指定し、第2引数で処理内容を指定する使い方をするようだ。
今後はdplyr::hogehoge_if, atを使わず、dplyr::acrossを用いるのが推奨されるようだ。
(ちなみに私は、可読性が多少下がるかもしれないがpackage::functionの書き方が好みである。
どのパッケージのどの関数を使っているかを明示的にすること、それによって関数同士のconflictを避けることもできること、並列処理の際に多少楽になることが利点としてあげられる。
このあたりは別途記事にしてみたい)